AlphaFold2 for Cyclic Peptide Design - A Deep-dive into Generative Drug Design (1/2)
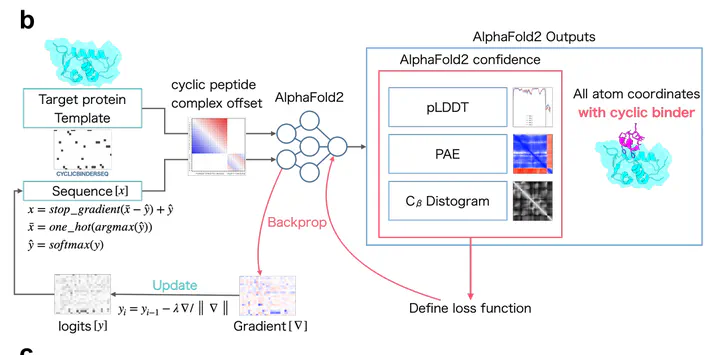 Cyclic Peptide generation architecture (Kosugi et. al.)
Cyclic Peptide generation architecture (Kosugi et. al.)As part of my learning journey on the use of AI for drug-discovery, I’ve documented my research notes, findings, and explorations into AlphaFold2 architecture and its use for de-novo cyclic peptide design. In this two-part series, I will first conduct a literature review and deep-dive into the AlphaFold2 model, and in part (2), I will explore recreating literature results and some exploratory hyperparameter searching.
Introduction
Predicting structural proteins is critical to the understanding of biology, developing novel therapeutics and innovating new compounds. In the past, these structures have been determined by experiments, but have not been able to cover the billions of known protein sequences. Google’s Deepmind’s AlphaFold offers a groundbreaking model that can accurately predict the protein structures close to experimental values in-silico, and has been a game-changer in the field of peptide design.
Researchers have recently adapted AlphaFold models to achieve designing of cyclic peptides. This project explored the use of AlphaFold and supporting bioinformatic tools to design cyclic-peptides, and is done so in three sections.
AlphaFold Architecture
AlphaFold uses a deep-learning architecture that uses two main stages - the Evoformer and Structure module (Fig 1). The Evoformer stages consist of 48 layers of Evoformer blocks and the structure module consists of 8 layers of the structure module block. AlphaFold uses an ensemble based approach of up to 5 models during inference to predict the structure of the sequence and its corresponding per residue local confidence (pLDDT), distogram, and predicted alignment error (PAE).
Input The model first receives as input the multiple sequence alignment (MSA), structural templates, and initial pair representations with positioning embeddings. The MSA is a database of protein sequences that have similar sequences across multiple biological forms (i.e. a human, rat, horse, etc.). The use of MSA is determined by the concept of coevolution, which states that changes in protein sequences will cause a complementary change by another sequence to maintain the overall protein structure. Another key input to the model is the position embedding to the distogram representation, the default positioning embedding used by AlphaFold is a linear and sequential embedding.
Evoformer block: The Evoformer then receives the input and performs a row-wise gated self-attention that is modified to add a bias from the pairwise representation to update the MSA row, followed by a column-wise gated self-attention to update each MSA column. The output is then constrained by a triangle inequality that ensures the geometric attributes of a molecular backbone. The triangle inequality ensures that a relationship is encoded amongst 3 connecting pairs of nodes based on their outgoing and incoming relationsips. Whilst a row attention is commonly used in other NLP transformers, the introduction of a columnar attention is a unique addition and allows the Evoformer to search through the “coevolution” spots in the MSA. By allocating query, key, value weights based on the importance of certain repeating or varying amino acids across the same MSA column, the Evoformer is able to pick-up on the patterns and variances in similar protein structures across organisms.
Structural block The structural block takes in as inputs the abstract pair-wise representation after the Evoformer and computes the geometric representation of the structure, confidence and loss of its expected prediction. The fundamental method AlphaFold uses to represent the molecular structure is by envisioning the backbone as a series of triangles, which are represented as a tuple of rotational and translational parameters from an initial frame of reference. The rotational and translational parameters for each backbone triangle is initialized at the center of the frame of reference, and the backbone is updated based on an attention mechanism between Evoformer outputs. To predict the sidechains, an additional neural network was trained to predict the torsion angles inspired to mimic and predict the Ramachandran plot, resulting in the final predicted structure.
ColabFold & ColabDesign: ColabFold is an adaptation of AlphaFold designed to provide a more accessible program for the broader community to use AlphaFold. Hosted on Google Colab, it provides a streamlined interface for protein structure prediction, a MSA retriever, and the ability to use Google Colab compute resources. The main method to use AlphaFold in this project is the use of Google Colab compute units [4].
ColabDesign is an adaptation of the ColabFold module to generate sequences that conform to a specific target structure. Based on the AlphaFold2 outputs, a loss function is defined based on pLDDT, PAE, and distogram to perform backpropagation through the AlphaFold2 model with frozen weights. In every pass of the generation process, the pairwise representation and amino sequence propagated to the AlphaFold network to generate the distogram and confidence scores. Whilst backpropagation based on the final structure is preferred, literature and implementation findings suggest a nonoptimal result: “28Feb2022 - We find backprop through structure module to be unstable, all functions have been updated to only use distogram by default. The definition of contact has changed to minimize entropy within distance cutoff.” [5] . Hence only the Evoformer output is used in this module and not the structural module in all ColabDesign. However, optimizing for the distogram will also optimize the final structure design, hence the results generated are still accurate.
Through a 3-stage approach, a gradient of logits for the sequence values are first changed, then the gradient descent is switched to being applied to the softmax output, and lastly to the one-hot-encodings. The gradient step changes for each of these stages, and ColabDesign has various hyperparameter options to adjust how the gradient descent is conducted:
Figure 5: ColabDesign gradient descent methods [7]
Cyclic Peptide Design and Prediction
The main method to design cyclic peptides lies in modifying the linear, sequential positional embeddings in the ColabDesign inputs, to enforce a circular relationship in how a cyclical structure is represented on the distogram. In the context of a cyclic peptide complex, this is represented in Fig.6 below in which the distance max is first determined to be the middle point of the sequence, followed by the minimum index distance between the starting residue and the ending residue, which is represented as a negative number.
Figure 6: Positional embedding comparison [8]
References
[1] Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., Petersen, S., Reiman, D., Clancy, E., Zielinski, M., Steinegger, M., Pacholska, M., Berghammer, T., Bodenstein, S., Silver, D., Vinyals, O., Senior, A. W., Kavukcuoglu, K., Kohli, P., & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
[2] Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S. A. A., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Silver, D., … & Hassabis, D. (2021). Supplementary information, highly accurate protein structure prediction with AlphaFold. Nature. https://doi.org/10.1038/s41586-021-03819-2
[3] Bouatta, N. (2023, February 16). Special lectures on machine learning and protein folding: Lecture 2. YouTube. https://www.youtube.com/watch?v=ri39B0Voujc&t=3769s
[4] Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., & Steinegger, M. (2022). ColabFold: making protein folding accessible to all. Nature Methods, 19(6), 679–682. https://doi.org/10.1038/s41592-022-01488-1
[5] https://github.com/sokrypton/ColabDesign?tab=readme-ov-file
[6] Rettie, S. A., Campbell, K. V., Bera, A. K., Kang, A., Kozlov, S., De La Cruz, J., Adebomi, V., Zhou, G., DiMaio, F., Ovchinnikov, S., & Bhardwaj, G. (n.d.). Cyclic peptide structure prediction and design using AlphaFold. University of Washington and Harvard University.
[7] https://github.com/sokrypton/ColabDesign/blob/main/af/README.md
[8] Takatsugu Kosugi and Masahito Ohue, Design of Cyclic Peptides Targeting Protein–Protein Interactions Using AlphaFold.